Similarity
Selection
Drop files to upload (or click)
About
SAXS Similarity generates a structural similarity map by comparing small-angle X-ray scattering profiles. There are a variety of metrics one can use to generate the matrix. For more information see Hura et al., 2013. The site is supported and brought to you by IDAT and the DOE.
Instructions
Updates 05/16/2022:
- A bug where the Rg and Scatter Plot would not show if a User clicked a square on the matrix plot for certain datasets is now fixed
- Filenames can now have consecutive dashes ('--'). Previously, this led Volatility of Ratio to fail
- The number of files you may upload has been increased from 60 to 100
- File extensions may now be '.txt' in addition to '.dat', however the filname preceding the extension must be unique (fix coming soon)
- File contents may now include headers starting with '#'
- Columns may be separated by any amount of white space (spaces or tabs)
- Lines may now end with DOS encoded CRLF line endings, i.e. those that contain Carriage Return (ASCII 13) characters
Files:
- Files must be plain text
- The file base name must consist of alphanumeric characters, dots (periods), dashes or underscores
- The entire filename must be 48 characters or less
- The file extension must be '.dat' or '.txt'
- Example: 'detector.test-file_0001.dat'
Data:
- The first column should contain the q-space (inverse Angstroms)
- The second column should contain the intensities
- The third column (optional) should contain errors
- Each datum should be represented as a float (includes exponential numbers)
- Columns should be separated by white space (spaces or tabs)
- Headers may be included if they start with a '#' hashtag/pound sign. Otherwise, there should be no other text within the files
- The default q range analyzed is 0.015 - 0.2, thus each data set must at least include this range
How to Upload:
- Drag and drop files on the tan box, or click the tan box to upload SAXS data files as a set
- You may upload up to 100 files
- The ability to add more files on top of the existing set is currently deprecated
Order of Files:
Files will initially be ordered based on the order they are uploaded. Once uploaded, the list of files displayed to the left of the color matrix can be rearranged by clicking and dragging files to their desired location within the list.
A number of automated sorting procedures are available under the 'Operations' tab near the top of the page. 'Fast Sort' uses a greedy algorithm to quickly sort the files based on their similarity. 'Slow Sort' uses a branch and bound algorithm to exhaustively search for the best file arrangement. This method should not be used for datasets greater then 9 files, as the time to complete the algorithm becomes inconvenient. 'Reverse Order' will vertically flip the matrix.
Further Analysis:
Once you have created an SSM, you may click on any matrix cell to report on the difference in Radius of Gyration as well as see a logarithmic plot of the two profiles. A slider bar under the Intensity plot can used to adjust the analyzed q range.
Different similarity metrics can be selected under the 'Method' tab near the top of the page: Volatility of ratio (default), Chi squared, Pearson Coefficient and Sokolova method.
The color range can be changed under the 'Color range' tab.
All the Rg differences between file pairs can be displayed or hidden within the color matrix by selecting 'Show/Hide Rg's' under the 'Operations' tab.
'Show Metric Matrix' found under the 'Operations' tab will display the raw values of the SSM.
'Convert to Image' also found under the 'Operations' tab will display the SSM and the color key as a downloadable png. For some browsers, the pop up blocker must be turned off.
Force Plot:
Another visualization tool, the force plot, displays each SAXS profile as a circular node. The size of the node is proportional to its Rg. The pairwise distances between nodes is proportional to similarity of the two files, described by the selected similarity metric. Nodes that are close together are more similar then nodes that are farther apart. Changing the similarity metric or the analyzed q range will update the force plot.
Example Data Set
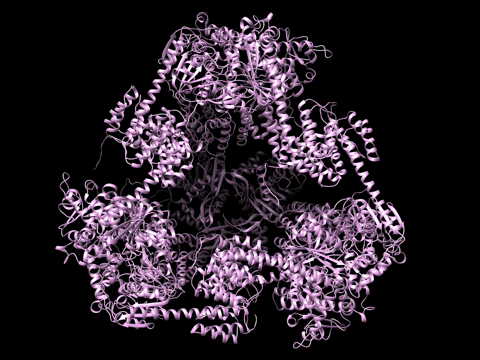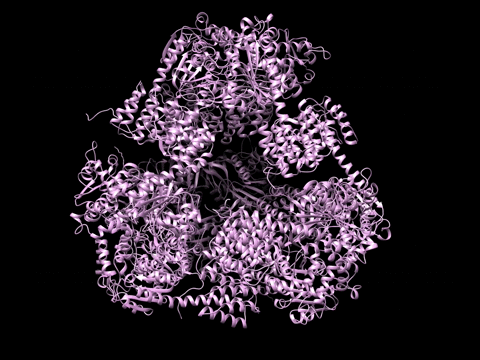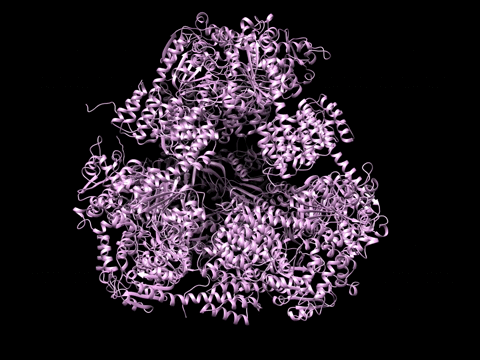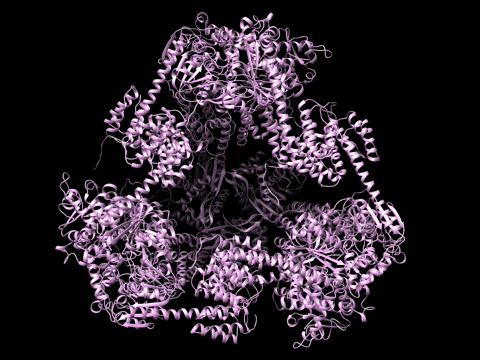 Example 1 Download the example data set below:
SAXS Similarity Maps (SSMs) are a way of comparing conformational states or structures. This data set is composed of calculated profiles from the morphed trajectory on the left. The files are named with a random start variable. However when loaded and clustered, they self organize according to the proper frame order as needed to construct the trajectory. Apply the Force Plot option and again they self organize according to Frame number. Note the diameter size of nodes in Force Plot is proportional to Rg.
Example 1 Download the example data set below:
SAXS Similarity Maps (SSMs) are a way of comparing conformational states or structures. This data set is composed of calculated profiles from the morphed trajectory on the left. The files are named with a random start variable. However when loaded and clustered, they self organize according to the proper frame order as needed to construct the trajectory. Apply the Force Plot option and again they self organize according to Frame number. Note the diameter size of nodes in Force Plot is proportional to Rg.
Example 2 Download the example data set below: This data set explores experimental data from the structure shown in Example 1. Data were collected as a function of salt (NaCl in mM 10, 100, 300, 500) vs pH (4-11). When first loaded, a non-intuitive pattern appears. Apply the clustering and note that all high pH structures cluster. At high pH (10 - 11) the assembly forms large heterogenoeus macromolecular assemblies. View in Force Plot and see the same cluster obviously seperates from the rest and that these nodes are larger diameter as diameter is set by Rg. Also investigate the other cluster in the Force Plot. The other cluser is mostly the same multimeric state shown in Example 1 and node separation is due to differences in conformation. At low salt the structure is compressed at high salt it expands.
Combine Example 1 and 2 for more insight into the experimental data
Example 3 Download the example data set below: The example compares ribosomal structures. Sorting ribosomal PDBs for comparison is challenging. Ribosomes are large and contain both protein and RNA making superposition for comparison challenging for most metrics. Ribosomal PDB entries are confusing as the large and small subunit must be stored as separate files. The example compares 24 recently added (10/1/2012) large subunits entries in the PDB of the ribosome through calculation of SAXS profiles. The resulting SSM allows one to sort the large and small subunit entires by comparison. 3TVE and 3TVF are the large and small subunits respectively.
Contact
For information about the app contact glhura@lbl.gov